FDA: Philips CPAP, BiPAP Recall Notification Inadequate

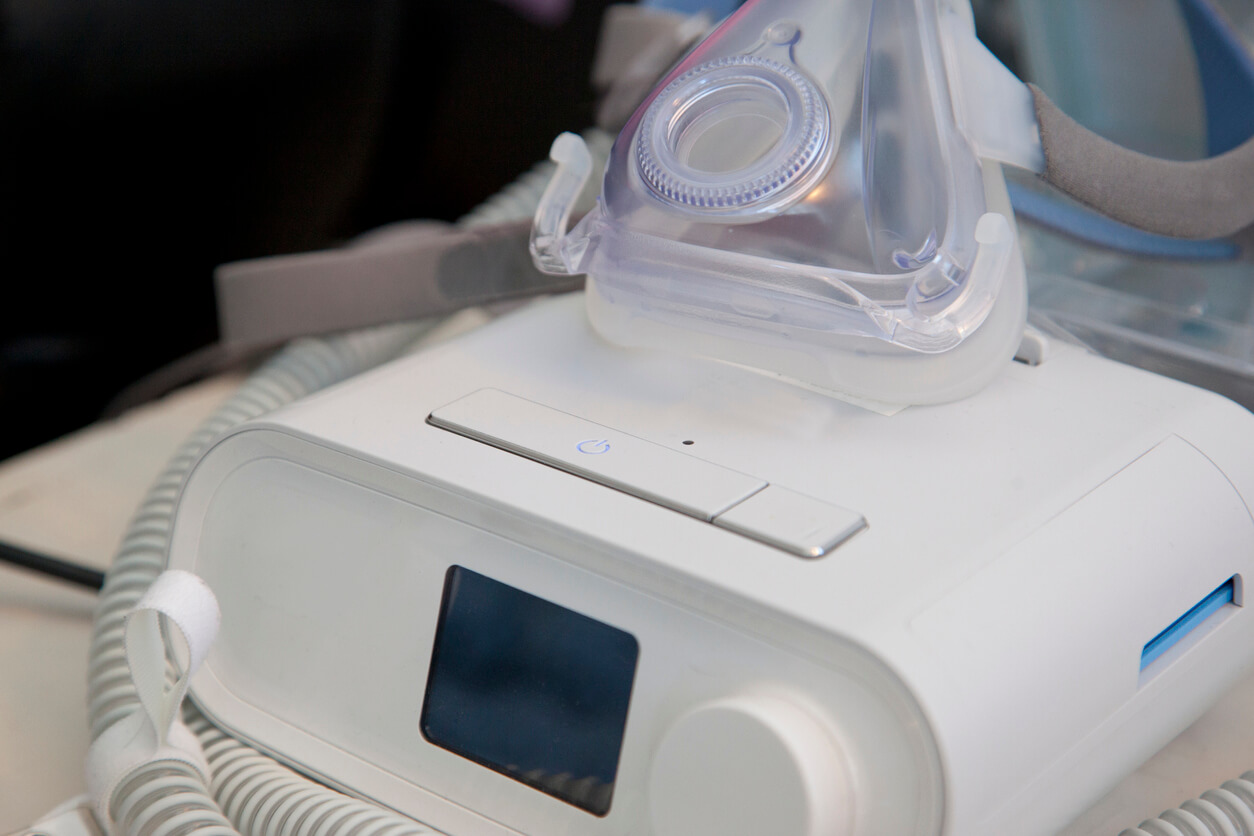
A notification order issued March 10 by the U.S. Food and Drug Administration to Philips Respironics said the company inadequately notified consumers and health professionals about a June 2021 recall of its CPAP, BiPAP and ventilator machines.
The FDA estimated that after more than nine months of recall communications from Philips, only 50% of people with the recalled devices had registered with the company to receive a replacement device.
Philips announced the voluntary recall of some models of continuous positive airway pressure (CPAP), bilevel positive airway pressure (BiPAP) and continuous ventilator devices on June 14, 2021. The recall was issued because the polyester-based polyurethane (PE-PUR) sound abatement foam used to reduce sound and vibration may break down and enter the device’s air pathway.
“The FDA has heard the frustration expressed by patients and durable medical equipment suppliers who are unaware of the recall and have received insufficient information on their next steps regarding the recall process,” said Jeff Shuren, M.D., J.D., director of the FDA’s Center for Devices and Radiological Health.
“Taking this action today enables the FDA to mandate that Philips Respironics improve its communication about the recall and the serious risk posed by the foam used in the recalled products with patients and the public and to ensure that individuals who rely on these essential devices are receiving the important information they need from the company.”
The FDA is requiring Philips to take specific actions to notify health professionals, distributors, retailers and device users of the recall within 45 days of the March 10 order.
Recommendations to Improve CPAP, BiPAP Recall Communication
The FDA recommends that Philips deliver monthly updates to users who register their devices on the Philips website. It asked Philips to give device users an expected time for replacement and provide the current rate of replacement for recalled devices.
In addition, the FDA recommended that Philips issue detailed information on the replacement process to device users, suppliers, distributors, retailers and prescribing health care providers.
Philips was also asked to provide online access to all available testing results. The FDA said the currently available information on the company’s website is vague and doesn’t provide the facts necessary for health care providers to make informed recommendations for patients using the recalled devices.
CPAP, BiPAP Recall History
In November 2021, the FDA updated the recall because it found a safety issue with the replacement foam Philips used on the recalled devices.
The agency discovered the replacement PE-PUR foam can degrade and emit volatile organic compounds, also known as VOCs. These VOCs can cause side effects, including breathing problems and irritation to the eyes, nose and throat.
Philips voluntarily initiated the recall because risks associated with the original PE-PUR foam could result in serious injury that may be life-threatening, cause permanent impairment or require medical intervention.
The foam has the potential to degrade and emit particles and toxic chemical gases into device air pathways that when inhaled can cause organ impairment, irreversible harm to lung tissues and long-lasting respiratory dysfunction. Potential risks from off-gassing include headaches, asthma, irritation, hypersensitivity, nausea, vomiting, dizziness and possible toxic and carcinogenic effects.
These health effects have led Philips CPAP owners to file individual lawsuits and several class-action lawsuits demanding financial damages.
Evidence shows Philips was testing alternative foams in 2016 that better resisted heat and humidity, which the company now says caused degradation of the original foam. Philips reportedly knew as early as 2015 that the PE-PUR foam had a degradation issue.