After 10 Years, the FDA Is Still Letting Women Down
Ten years after our initial report on how the U.S. Food and Drug Administration's shortcomings directly impact women, the agency is still under scrutiny. The FDA has made progress by including more women in clinical trials, but its medical device and drug regulations still fall short, leading to injuries and lawsuits.


In Drugwatch’s 2015 investigation, How the FDA Let Women Down, we highlighted issues with drug and medical device approvals that posed greater risks to women.
Now, we’re diving deeper into regulatory processes to highlight how far they’ve come — and where the administration still falls short in terms of device testing and clinical trials for medical products marketed toward women.
The FDA’s 510(k) clearance process is still allowing moderate-risk devices on the market without clinical trials. Some of these products, such as pelvic mesh, continue to hurt women.
The agency has also been working to approve drugs faster than ever, offering fast-track options for new drugs for serious illnesses such as cancer.
However, mistakes can lead to devastating outcomes when drugs are approved based on lower-quality data. In some cases, the FDA proposed using one clinical trial with patients instead of two to approve drugs faster.
More recently, the FDA has championed AI to help achieve faster drug approvals, but AI has been known to produce false data.
As health care evolves, so do women’s needs and safety concerns. Stronger data and testing requirements can help protect women from dangerous medical devices and drugs.

Medical Approval Processes May Fall Short
While the FDA requires clinical trials for drugs to hit the market, a large number of medical devices are sold without trial data — exposing women and men to health risks.
The 510(k) clearance process allows medium-risk (Class II) medical devices like surgical mesh, some hip implants, catheters, pregnancy tests and others on the market without clinical trials as long as they are similar to devices already on the market.

Drug approvals, on the other hand, require more testing and clinical data for the FDA to approve them. However, in some cases, the quality of the data submitted may be an issue, and drugs could be approved based on lower standards.

Medical Devices: Inadequate Testing, Conflicts of Interest and Delayed Warnings
Donna Miser’s doctor implanted a surgical mesh bladder sling that was supposed to help her with stress urinary incontinence (SUI), a condition that causes urine to leak when there’s increased pressure on the bladder. Exercising, sneezing or coughing can all trigger these leaks. SUI affects 1 in 3 women.
But no one told her about the risks of mesh.
The implant is supposed to be permanent, but after a few years, the mesh eroded into her bladder and vaginal walls and cut into her urethra in multiple places. She’s since had several surgeries to remove the mesh.
“Someone’s really dropped the ball. I do not understand how so many women got implanted with [this] product. That surgeon looked at me with a smile on his face, telling me, ‘I have got the answer. I’ve got the cure for you. We’re going to put this in you,’” Miser told Drugwatch. “It wasn’t tested. It wasn’t approved.”
510(k) Medical Device Approval Leaves Gaps
When Miser said her mesh wasn’t tested or approved, she wasn’t wrong. The 510(k) process allows devices to be approved without clinical trials if they are similar to products already on the market, which are called predicate devices.
The issue with 510(k) approvals is that products can enter the market based on similarities to decades-old devices. This was the case with the surgical mesh implanted in women for SUI or pelvic organ prolapse (POP), a condition where organs slip down and bulge into the vagina.
Another, more rigorous (but less frequently used) path to device approval, Premarket Approval (PMA), requires more scientific evidence. The PMA is intended for high-risk Class III medical devices, such as pacemakers or defibrillators.
Mesh implanted through the vagina for POP has since been reclassified to a Class III device and now requires more testing before it’s sold, but SUI mesh remains a Class II.
“Someone's really dropped the ball. I do not understand how so many women got implanted with [this] product.”
Some Devices Were Approved Based on Recalled Devices
Some of those predicate devices used for the 510(k) process may have been recalled for safety reasons. However, products that were allowed on the market based on these devices aren’t being recalled.

For example, researchers from the Centre for Evidence-Based Medicine at Oxford University traced 61 transvaginal mesh products based on two predicate meshes, Mersilene Mesh (approved in 1985), and the ProteGen Sling (approved in 1996). The ProteGen Sling was recalled in 1999 for safety reasons, but products based on the ProteGen remained on the market.
Pelvic mesh devices have caused serious, debilitating complications such as pain, bleeding, inflammation and infections in many women like Donna Miser. Mesh implants have been at the center of countless lawsuits. Transvaginal mesh cases alone have led to roughly $8 billion in settlements for around 100,000 women.
510(K) Fees Fund FDA Reviews
Another issue brought up by critics of the 510(k) program is that medical device companies pay fees that fund the FDA’s review process. This could potentially lead to conflicts of interest.
“The 510(k) is the workhorse of industry at the FDA because 510(k) essentially gives greenlights to a lot of products without any demonstration of safety and efficacy.”
Medical device manufacturers pay fees to the FDA along with their applications. These fees fund the FDA review process. In 2025, the standard 510(k) fee is $24,335, while the more rigorous device approval path, the PMA, costs $540,783.
“The 510(k) is the workhorse of industry at the FDA because 510(k) essentially gives greenlights to a lot of products without any demonstration of safety and efficacy,” according to Dr. Hooman Noorchashm, a research professor at Northeastern University School of Law and co-director of the Amy J. Reed Medical Device Safety Collaborative.

The FDA and its Center for Devices and Radiological Health (CDRH) are responsible for ensuring medical devices and radiation-emitting products are safe and work appropriately.
Industry funding of the FDA and CDRH can raise concerns over how these regulations are enforced, according to Noorchashm.
Missing Safety Device Data May Delay FDA Warnings
The FDA’s Manufacturer and User Facility Device Experience (MAUDE) system is a searchable database for medical device complications. The FDA uses it to flag safety data and determine if it needs to take action.
Madris Kinard of Device Events used to work at the FDA as an adverse events subject matter expert for devices and unique device identification (UDI). Kinard spoke to Drugwatch and cited a report on a problematic birth control device called Essure. With Essure, doctors implanted two metal coils into the fallopian tubes. This caused scar tissue to develop, blocking the tubes and preventing sperm from reaching the egg.
Women reported thousands of complications from Essure that led them to get it surgically removed.
Kindard’s FDA database analysis showed that about 32,000 device complaints from inspections of Essure’s manufacturer in 2011 and 2013 hadn’t made it into the FDA’s database. Kinard said it’s not clear whether these complaints contained adverse event reports because the details haven’t been made public. The FDA still hasn’t responded to her Freedom of Information Act (FOIA) request.
If that data had been added to MAUDE, it might have given the FDA more information to warn women about Essure sooner.
“That set them back by probably 10 years in identifying these problems,” Kinard said.
Ten years ago, Dr. Noorchashm and his wife, Amy Reed, fought to get a medical device called a power morcellator removed from the market.
Doctors used the device, approved through the 510(k), to remove Reed’s uterine fibroids. It was supposed to be a safe, minimally invasive procedure. Instead, the morcellator spread cancer cells hidden in the fibroids throughout her abdominal cavity. Three years later, she died at 44.
Because of Noorchashm and Reed’s advocacy, the FDA severely restricted power morcellator use and added a black box warning to the device.
Maria Gmitro is an FDA consumer representative and founder of Breast Implant Safety Alliance (BISA), a patient advocacy group focused on implant safety. Because of the work of BISA and the patient advocates, the FDA strengthened breast implant safety requirements — including restricting the sale of implants, adding a boxed warning and providing a patient decision checklist.
“The checklist was created because patients were not warned. And advocates had to push for that. Not the plastic surgery society, not industry, but patient advocates had to push for that,” Gmitro told Drugwatch. “Survivors made that happen.”
Expert Opinion: How Can the FDA Improve 510(k) Medical Device Regulation?
In 1976, regulators didn’t intend the 510(k) to be the dominant way for medical devices to make it to market. For that to be remedied, CDRH needs to take steps toward reform, Noorchashm said.
“The [FDA’s] real job is not to be an arm of industry. Its real job is to make sure that products are safe, and the market can deliver adequate signals,” he added.
He suggested that the FDA continue to allow the industry to use 510(k), but let the public know that it’s not designed to prove safety and efficacy. Then, it will be the court’s job to evaluate products when litigation is necessary.
In 2018, the FDA announced it was going to modernize the 510(k), which has been in place since 1976. The agency released draft guidance on how to properly select a predicate device. The original should:
- Be cleared using well-established methods
- Meet or exceed expected safety and performance
- Not be associated with a design-related recall
- Not have unmitigated use-related or design-related safety issues
It remains to be seen if the FDA intends to do more, as the recommendations aren’t mandatory.

Drugs: Poor Evidence for Approval, Improper Doses for Women and Underrepresentation in Clinical Trials
Unlike the 510(k) clearance pathway for medical devices, drugs require more clinical trial data for approval. One of the most important parts of the drug approval process is when the FDA looks at the risks and benefits from clinical trial data submitted by a manufacturer. The FDA expects the manufacturer to conduct two well-designed clinical trials, but in some cases, it will accept one.
To determine if drugs work safely, the FDA uses four minimum criteria to judge whether manufacturers have provided enough evidence for drug approval.
- Control group: People who took the drugs in clinical trials were compared to a control group. People in a control group take a placebo or other form of care but don’t receive the drug being studied.
- Blinding: Neither clinical trial subjects nor doctors know which patients are taking the actual drug, a placebo or another treatment.
- Replication: There should be at least two “well-controlled” trials showing the drug worked.
- Clinical endpoint: Studies are based on clinical endpoints. These endpoints measure the drug’s effect on how a patient feels, functions or survives — the most reliable outcomes — rather than a surrogate measure that only predicts these effects, such as a blood test or CT scan.
A new report from The Lever and the McGraw Center for Business Journalism at CUNY’s Newmark Graduate School of Journalism analyzed a government database and looked at 429 drugs approved by the FDA from January 2013 to December 31, 2022.

The report revealed that the FDA approved these drugs without clinical trials that met the minimum four criteria of having a control group, blinding, replication or clinical endpoints.
“More medical products have been allowed on the market in the last decade based on skimpier research, or research studying biological markers that patients can’t feel (such as plaques on the brain or bone density) rather than meaningful health benefits such as living longer or spending less time in a hospital,” Diana Zuckerman, President of the National Center for Health Research and a project advisor for The Lever, told Drugwatch.
| Drug | Used to Treat | Control Group | Blinded | Replicated | Clinical or Surrogate Outcome |
|---|---|---|---|---|---|
| Elahere | Ovarian cancer | No | No | No | Surrogate |
| Enhertu | Breast cancer | No | No | No | Surrogate |
| ExEm Foam | Assess fallopian tube openness in known or suspected infertility | Yes | Yes | No | Surrogate |
| Ibrance | Advanced breast cancer | Yes | No | No | Surrogate |
| Jemperli | Endometrial cancer | No | Yes | No | Surrogate |
| Kadcyla | HER2+ late-stage breast cancer | Yes | No | No | Clinical |
| Kisqali | Advanced breast cancer in postmenopausal women | Yes | Yes | No | Surrogate |
| Lynparza | Advanced ovarian cancer | Yes | Yes | Yes | Surrogate |
| Margenza | HER2+ breast cancer | Yes | No | No | Surrogate |
| Nerlynx | Reduce the risk of breast cancer coming back | Yes | Yes | No | Surrogate |
| Nextstellis | Prevent pregnancy | No | No | No | Clinical |
| Ozempic | Type-2 diabetes and off-label for weight loss | Yes | Yes | Yes | Surrogate |
| Piqray | Breast cancer | Yes | Yes | No | Surrogate |
| Recarbrio | Complicated UTIs and intra-abdominal infections | No | No | No | Surrogate |
| Rubraca | Ovarian cancer | No | No | Yes | Surrogate |
| Tukysa | Unresectable, metastatic HER2+ breast cancer | Yes | Yes | No | Surrogate |
| Verzenio | Advanced, metastatic breast cancer | Yes | Yes | No | Surrogate |
| Zejula | Ovarian, fallopian tube or primary peritoneal cancer | Yes | Yes | No | Surrogate |
| Zemdri | UTIs | Yes | Yes | No | Clinical |
Investigative journalists Jeanne Lenzer and Shannon Brownlee spearheaded the database project and found several surprises in the data.
“We knew going in that the FDA had relaxed its scientific standards over the years and that the result was drugs getting on the market without adequate evidence that they work,” Lenzer told Drugwatch. “We didn’t know just how bad it was.”
Lenzer and Brownlee were also surprised by how many cancer drugs in the data they pulled made it to the market without adequate proof they work. The exact cancer medications are included in the table above.
Improper Dosing Can Lead to More Side Effects For Women
Women experience side effects nearly twice as often as men, and one of the reasons is that medications take longer to leave women’s bodies.
Even with researchers recommending dose reductions for women, the FDA hasn’t taken meaningful action to require sex-specific dosing information on drug labels.

One study in Biology of Sex Differences looked at 86 drugs and found that (when compared to men), women generally had higher blood concentrations of the drugs, and the medications took longer to leave their bodies. This led to higher rates of side effects in women in 96% of cases.
The findings in this study suggest that women may have been prescribed higher doses of drugs than necessary, even when they take the dose recommended on the drug’s label or as directed.
Medications studied included common OTC and prescription medications such as aspirin and Zoloft (sertraline).
| Generic Drug Name | Side Effects |
|---|---|
| Liraglutide | Headache, vomiting, nausea |
| Rosiglitazone | Fracture |
| Heparin | Blood disorders, bleeding, lymphatic disorders |
| Warfarin | Bleeding |
| Torasemide | Hospitalization |
| Pravastatin | Coronary heart disease |
| Amlodipine | Edema, palpitations, flushing |
| Digoxin | Death |
| Trospium | Cognitive impairment |
| Prednisone | Hair loss, mood swings, weight gain, depression, fatigue |
| Levofloxacin | Fluoroquinolone toxicity |
| Paclitaxel | Death, myocardial infarction, lesion, revascularization |
| Infliximab | Allergic reactions |
| Adalimumab | Allergic reactions |
| Propranolol | Dry mouth, muscle pain, headache, dizziness |
| Oxycodone | Nausea, skin itching |
| Buprenorphine | Sleep disturbances |
| Carbamazepine | Cognitive impairment, elevated cholesterol |
| Gabapentin | Nausea, dizziness, excessive sleepiness |
| Aripiprazole | Blood pressure, elongated QTc |
| Risperidone | Neurological effects, headache, low blood pressure |
| Fluoxetine | High cholesterol, suicidal ideation |
Older Women and Women of Color Still Underrepresented
When we interviewed Zuckerman in 2015, she highlighted the lack of women, people of color or people over age 65 in clinical trials. Over the past 10 years, the FDA has increased the number of women in clinical trials, but still lags behind with women of color and older women.
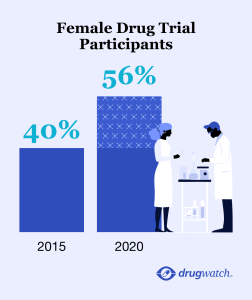
“Most trials submitted to the FDA include quite a few women, but they are not women of color or women over 65, even though many diseases are more common on people over 65 and at least as prevalent in people of color,” Zuckerman told Drugwatch.
While it’s great that more women are finally included in trials, the benefits might not be seen for a few years. Most drugs on the market today were approved during older clinical trials. The data from these trials were primarily gathered from men, leaving a gap in safety data for women.
Using AI to Speed Up FDA Reviews Could Lead to False Information
FDA Commissioner Dr. Martin Makary is leading the charge to integrate generative AI into FDA reviews. While there’s no doubt AI can help speed up workflows, it’s also known to generate false data and phantom citations.
The Make America Healthy Again (MAHA) Commission report has been cited by experts as an example of how AI can conjure false data, though the Department of Health and Human Services hasn’t admitted to whether it used AI in the report’s creation. The report contained multiple mistakes and phantom citations.
American Public Health Association Executive Director Dr. Georges Benjamin told the Washington Post that the report can’t be used for policymaking or discussion because it’s unreliable. Diana Zuckerman shares the same sentiment.
“The bottom line is that current AI technology can’t be trusted to be accurate, so whatever information it provides needs to be checked by humans, and that can add time as well as save time,” Zuckerman told Drugwatch. “As drugs and biologics become more complex, the FDA needs more people with a range of expertise that wasn’t needed before, and at this point AI can’t replace those types of scientific experts.”
Expert Opinion: How Can the FDA Improve Drug Safety?
When it comes to drug safety, the FDA needs to require more stringent clinical trial evidence before allowing drugs onto the market.
“The problem for the agency is it is now hamstrung by Congress, which has, over the years, steadily eroded the statutes governing drug regulation,” Lenzer said. “In addition, we believe that PDUFA has to be repealed.”
The PDUFA, or Prescription Drug User Fee Act, allows the FDA to collect fees from drugmakers in exchange for expediting their medications’ reviews and approvals.
“Nobody wants to talk about that because it would almost certainly require public funding, but an agency that is paid by the industry it is supposed to regulate — almost by definition — cannot be independent. What that means for the FDA is it no longer protects the public health and patients because it’s too busy protecting the commercial interests of its benefactors,” Brownlee added.

Government Funding Shouldn’t Be a Hindrance
At the end of the day, the FDA needs money to make changes to better protect women and the public. Budget cuts across government agencies might make that money tough to come by.
Dr. Elias Zerhouni served as the Director of the NIH from 2002 to 2008 and authored the upcoming book Disease Knows No Politics. He believes the FDA’s power to regulate should be maintained despite budget cuts and provided his insight as a scientist and researcher.
He brought up the Department of Government Efficiency (DOGE) oversights in recent months. The department let some FDA staff members go before rehiring them. Zerhouni said he has never seen such a drastic budget cut done so quickly at the Department of Health and Human Services.
“They closed a lot of websites. Now they’re reopening them. I think it’s a little chaotic. I hope cooler heads prevail soon … because I don’t think you can sustain that over time,” he said.
From Zerhouni’s experience, no one is aiming to harm the regulatory processes in the country — even in this administration. However, inexperience and political priorities related to reducing budgets are causing chaos. As co-founder of ModeX Therapeutics, a small biotech company, Zerhouni hasn’t seen a slowdown in the responsiveness of the FDA.
However, whether it should be or not, women’s health is political, and politics shouldn’t stand in the way of progress.
“Let’s not forget that women’s health is a prisoner — is a hostage of political divisions in this country,” said Zerhouni.

Notable Women's Health Lawsuits
Women have filed lawsuits after being injured by several health-related products allowed on the market by the FDA. We’ve highlighted some current litigations that affect women, including what the FDA has done regarding the safety concerns raised in the lawsuits.
Allergan Breast Implants and Cancer
Allergan breast implant lawsuits claim Allergan’s Biocell textured breast implants triggered a type of cancer called breast implant-associated large cell lymphoma (BIA-ALCL). According to the data, most BIA-ALCL (91%) cases happened in women with a Biocell implant.
In 2019, the FDA told Allergan to recall its Biocell implants and released safety communications warning the public. It also recommended changing implant labeling to include a boxed warning and providing patients with a checklist to better understand the risks.
Depo-Provera and Brain Tumors
A March 2024 study found that women who used the Depo-Provera birth control shot had a 5.55-fold increased risk of developing meningioma, a tumor in the lining of the brain.
Depo-Provera lawsuits claim drugmakers failed to warn the public of this risk. So far, the FDA hasn’t addressed this issue in any guidance or safety communications.
Hair Relaxers and Cancer
An October 2022 study in the Journal of the National Cancer Institute found that women who used hair relaxers at least five times in the previous year were more than twice as likely to develop uterine cancer.
As of August 2025, there were more than 10,000 lawsuits against hair relaxer makers, claiming the company’s relaxer products caused cancer.
The FDA hasn’t addressed the overall cancer risk of hair relaxer chemicals. Unlike drugs, the FDA doesn’t require trials to prove cosmetic products like hair relaxers are safe, and cosmetics don’t need FDA premarket approval. The FDA leaves the duty of ensuring product safety to manufacturers.
It did, however, propose a ban on formaldehyde — one of the cancer-causing chemicals addressed in the October 2022 study — in hair straightening products in 2024. But the ban was placed on hold indefinitely after an executive order paused all federal regulations.
Talc and Ovarian Cancer
There have been over 59,500 lawsuits against Johnson & Johnson for Johnson’s Baby Powder and Shower to Shower products as of June 2025. Women filed lawsuits claiming talcum powder in those products had been contaminated by asbestos, which may have caused ovarian cancer when used in the genital area for several years.
In 2019, the FDA alerted consumers that asbestos had been found in a sample of Johnson’s Baby Powder. J&J recalled the lot. In 2024, the FDA proposed a requirement for manufacturers to test for asbestos in talc-containing cosmetic products, but there has been no update on this.
Paragard IUD Breakage
Women have filed Paragard lawsuits because the birth control IUD’s arms may break when removed. Injuries include surgery to remove the broken device, infertility that persists after removal and pain.
The FDA quietly launched an investigation into reports of device breakage beginning in 2021. The FDA didn’t make its findings public.
In June 2024, the agency required Cooper Surgical, the IUD’s maker, to add more warnings about the IUD potentially breaking.
Power Morcellators and Cancer
Women filed power morcellator lawsuits after the device spread undetected cancer cells in their abdomens. This litigation is no longer active, but the main defendants were Johnson & Johnson and Karl Storz.
About 100 lawsuits were filed against Johnson & Johnson and the company ended up settling the cases for an undisclosed amount. Lawyers estimated the settlements ranged from $100,000 to $1 million.
Transvaginal Mesh Requiring Removal Surgery
Thousands of women have filed transvaginal mesh lawsuits against multiple medical device companies for complications such as infections, mesh cutting into other organs, bowel issues and more.
Like Donna Miser, many of these women required multiple surgeries to remove the mesh. In 2019, the New York Times reported that settlements neared $8 billion.
Even after some settlements have been reached, lawyers are still accepting cases.
Women who used transvaginal mesh for POP and SUI have filed lawsuits. While the FDA has reclassified POP mesh as a Class III device requiring clinical trials for approval, transvaginal mesh for SUI remains a Class II device cleared for sale through the 510(k).
Calling this number connects you with a Drugwatch.com representative. We will direct you to one of our trusted legal partners for a free case review.
Drugwatch.com's trusted legal partners support the organization's mission to keep people safe from dangerous drugs and medical devices. For more information, visit our partners page.