Knee Replacement Recalls
Knee replacement recalls have been issued because of loosening, early wear or faulty packaging. Nearly 1,000 implant parts used in knee surgeries have been affected by DePuy, Zimmer Biomet and Stryker knee replacement recalls. Lawsuits blame manufacturers for selling defective devices.
Our content is developed and backed by respected legal, medical and scientific experts. More than 30 contributors, including product liability attorneys and board-certified physicians, have reviewed our website to ensure it’s medically sound and legally accurate.
legal help when you need it most.
Drugwatch has provided people injured by harmful drugs and devices with reliable answers and experienced legal help since 2009. Brought to you by The Wilson Firm LLP, we've pursued justice for more than 20,000 families and secured $324 million in settlements and verdicts against negligent manufacturers.
More than 30 contributors, including mass tort attorneys and board-certified doctors, have reviewed our website and added their unique perspectives to ensure you get the most updated and highest quality information.
Drugwatch.com is AACI-certified as a trusted medical content website and is produced by lawyers, a patient advocate and award-winning journalists whose affiliations include the American Bar Association and the American Medical Writers Association.
About Drugwatch.com
- 17 Years of Advocacy
- $324 Million Recovered for Clients
- 20,000 Families Helped
- A+ BBB Rating
- 4.9 Stars from Google Reviews
Testimonials
I found Drugwatch to be very helpful with finding the right lawyers. We had the opportunity to share our story as well, so that more people can be aware of NEC. We are forever grateful for them.



- Medically reviewed by Jessica D. Hess, Ph.D.
- Last update: May 5, 2026
- Est. Read Time: 5 min read
- Knee Replacement Safety
- Reasons for Recalls
- Manufacturers With the Most Recalls
- DePuy Knee Replacement Recalls
- Zimmer Biomet Knee Replacement Recalls
- Stryker Knee Replacement Recalls
- Wright Medical Knee Replacement Recalls
- Arthrex Inc. Knee Replacement Recalls
- Exactech Transitions GXL Liners Out of the US Market
The advocacy group Consumers Union reviewed the U.S. Food and Drug Administration’s medical device database and found that 709 knee replacement recalls were issued between 2003 and 2013. Drugwatch conducted a separate review in 2018 and identified another 76 recalls. Roughly 97% of all recalls during that timeframe involved knee replacements from just three manufacturers: Zimmer Biomet, DePuy and Stryker.
Smith & Nephew recalled more than 42,000 Journey I BCS Knee Systems in 2018 after joint registries in the United Kingdom and Australia — which collect data on joint replacement surgeries to improve patient care — reported the device’s revision surgery rates were 1.5 times that of the average for all knee replacements.
Manufacturers and Knee Replacement Safety
Knee implant manufacturers are responsible for appropriate marketing of their products. They must warn consumers and health care professionals about their devices’ risks. In July 2022, one research study found that over 90% of patients have excessive sedentary behavior following total knee replacement surgery while nearly half experience clinically significant fatigue after five years.
Manufacturers are also responsible for monitoring their products for problems after FDA approval. But critics argue that the FDA approval process does not do enough to keep unsafe devices off the market. Manufacturers often rely on the agency’s Premarket Notification process. This is also called the 510(k) process. It lets knee replacement makers skip more rigorous tests. They only have to show their device is “substantially equivalent” to similar products.
Knee replacement lawsuits blame manufacturers for selling defective devices.
Reasons for Knee Replacement Recalls
Manufacturers issue knee replacement recalls for several reasons. These can be as simple as faulty packaging on knee replacement parts, or they can involve defective parts that pose a risk of serious injuries.
- Faulty design
- Implants and tools were prone to fracture. Tools had flawed designs or sometimes fell into the surgical site
- Improper fit
- Implants could suffer damage when surgeons forced a component into place
- Loosening
- Patients needed revision surgery because knee replacements loosened
- Early wear
- Parts wore out sooner than expected
- Packaging errors
- Faulty packages compromised surgical tools’ sterility
- Co-mingled components
- Parts of the left and right knee replacements became mixed together. This caused confusion during surgery

Manufacturers With the Most Knee Replacement Recalls
Three manufacturers account for the majority of knee replacement recalls since 2003. DePuy Synthes accounted for almost half of the total recalls. Two other companies combined for only 15 recalls between them.
| Knee Replacement Manufacturer | Knee Recalls |
|---|---|
| DePuy Synthes | 485 |
| Zimmer Biomet* | 355 |
| Stryker | 125 |
| Smith & Nephew | 15 |
| Wright Medical** | 4 |
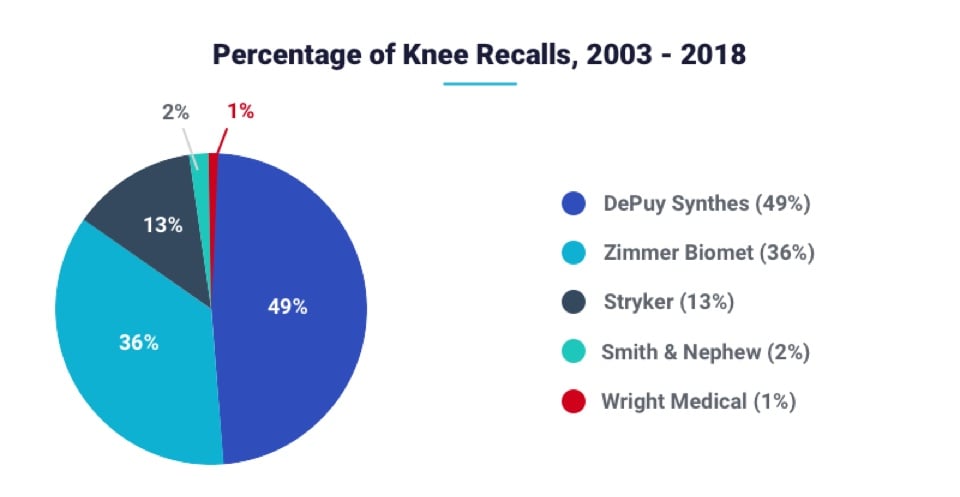
**Wright sold off its hip and knee replacement business in 2016. It no longer makes knee replacement implants.
DePuy Knee Replacement Recalls
DePuy Synthes has issued hundreds of recalls over the years. One of the most recent was in December 2017 for Sigma knee components. The devices had higher-than-expected failure rates. DePuy also recalled almost 14,000 Attune knee surgical tools in 2015. But to date, there have been no recalls of Attune Knee implants.
The FDA’s database shows at least six other DePuy knee replacement recalls since 2013. Between 2003 and 2013, DePuy issued at least 200 knee replacement-related recalls. That’s more than any other knee implant manufacturer.
| Date | Amount | Device Type | Reason |
|---|---|---|---|
| December 2017 | 7,500 | SIGMA HP PFJ Cemented Trochlear Implants | Elevated revision rates |
| January 2016 | 351 | SIGMA HP MBT Non-Keel Punch Knee Instruments | Design flaw that could cause a delay in surgery |
| September 2015 | 7,488 | Specialist 2 Intramedullary (SP2 IM) Rod | Instrument fracturing during surgery and leaving parts of the rods in patients |
| June 2015 | 13,964 | Attune Knee Tibial Articulation Surface Instruments | Could come off during surgery, leaving a part in device in the patient |
| March 2015 | 28,732 | LCS Complete RPS Inserts | Reports of higher revision surgery rates in Australia |
| November 2014 | 7,944 | Attune Intuition Impaction Handle | Reports of fractures potentially leaving parts in the patient |
| November 2014 | 4,555 | Attune Intuition Impactors | Could fracture and pieces could be left in the patient |
| February 2014 | 129 | S-ROM Noiles Rotating Hinges | Defective packaging could compromise sterility |
Zimmer Biomet Knee Replacement Recalls
Zimmer Biomet has issued at least 100 knee replacement-related recalls since 2003. One of the most recent was in 2017 for more than 28,000 joint components. Inspections found elevated endotoxin levels on the parts.
Zimmer Biomet is the largest knee-implant maker in the world. The company took on its present form with the merger of Zimmer and Biomet in 2015.
| Date | Units | Device Type | Reason |
|---|---|---|---|
| November 2017 | 13,227 | Gender Solutions Patello-Femoral Joint System | Packaging problems |
| November 2017 | 1023 | Persona Partial Knee instruments | Spacer blocks do not fit in the alignment tower instrument or handle |
| October 2017 | 430 | Persona Partial Knee Spacer Blocks | Problems with flexion in range-of-motion trials |
| February 2017 | 28,000 | Zimmer Biomet polyethylene joint components | Elevated endotoxin levels |
| January 2017 | 15,000 | Vanguard Total Knee System | Mislabeling and packaging problems |
| May 2016 | 10,256 | Persona Trabecular Metal Tibial Plate Knee implants | Implant loosening |
| January 2016 | 42,064 | Zimmer NexGen Knee implants | Packaging could adhere to implant parts |
Stryker Knee Replacement Recalls
There have been at least 125 Stryker knee replacement recalls since 2003.
Stryker voluntarily recalled its ShapeMatch Cutting Guides after patients reported injuries. These instruments were meant to help surgeons position the Stryker Triathlon knee implant before the bone was cut, but complaints submitted to the FDA allege the device was misaligned, which caused such complications as joint instability, chronic pain and revision surgery. The FDA later classified it as a Class I recall in April 2013.
In 2016, the company issued two recalls affecting 14,000 surgical instruments.
| Date | Amount | Device Type | Reason |
|---|---|---|---|
| August 2016 | 2,039 | Stryker Orthopaedics Patella Assembly Instruments | Parts could come apart from an accompanying instrument during surgery |
| June 2016 | 12,469 | Modular Handle Triathlon Instruments | Parts could come apart from the rest of the instrument |
| September 2015 | 3,444 | MIS Modular Distal Capture Triathlon Instruments | Reports that parts could come apart from the instruments |
| August 2014 | 1,147 | Triathlon Femoral Components | Packaging issue could affect the shelf life of the devices’ sterility |
| August 2014 | 1,147 | Scorpio Femoral Components | Packaging issue could affect the shelf life of the devices’ sterility |
| April 2013 | 7,868 | ShapeMatch Cutting Guide | Device misalignment could result in knee implants being placed in the wrong position |
Smith & Nephew Knee Replacement Recalls
Smith & Nephew has announced at least 15 knee replacement-related recalls since 2003. The 2018 recall of the Journey I BCS Knee System affected more than 42,000 units, according to the FDA.
- JOURNEY BCS CoCr Femoral Component
- JOURNEY BCS Oxinium Femoral Component
Previous Smith & Nephew recalls involved fewer than 100 total units.
| DATE | AMOUNT | DEVICE TYPE | REASON |
|---|---|---|---|
| June 2018 | 42,050 | Journey I BCS Knee System | Higher than normal revision rates because of early component loosening and other problems |
| May 2017 | 46 | Legion Tib Cone Impactor Heads | The wrong adhesive used in assembly |
| March 2016 | 24 | TC-Plus Primary Tibial Components | Manufacturing defect; it could make the polyethylene insert difficult or impossible to seat during surgery |
| January 2016 | 8 | Legion Hemi Stepped Tibial Screw-On Wedges | Screws were too long |
Wright Medical Knee Replacement Recalls
Wright Medical announced four knee replacement recalls since 2003. Drugwatch found none after 2013. Wright sold its hip and knee division to Corin Orthopaedics in 2016 for a reported $290 million. Wright is no longer a major player in the knee-implant business.
Arthrex Inc. Knee Replacement Recalls
Arthrex has issued two recalls related to its iBalance knee replacement line: one for the iBalance Total Knee Arthroplasty System and another for the iBalance Unicondylar Knee Arthroplasty System.
| Date | Amount | Device Type | Reason |
|---|---|---|---|
| December 2015 | 2,378 | Arthrex iBalance TKA Tibial Tray | Outer metal surface was smooth compared to previous textured components |
| October 2017 | 20 | iBalance(R) UKA, Femoral Cemented, Size 3, Left Medial/Right LATERAL | Incorrect part number printed on label |
Exactech Transitions GXL Liners Out of the US Market
While Exactech has noted that it is not officially recalling its Connexion GXL hip replacement liners, in letters provided to surgeons and patients in June 2021, the company explained that GXL liners were transitioned out of the US market due to risks of premature wear and possible complications.
The company stated: “No, Exactech is not recalling the GXL liner. Overall, the GXL Acetabular liner is considered safe and effective and performs as intended. However, in March 2018, Exactech received FDA approval for our next generation of polyethylene highly crosslinked Vitamin E liners, XLE. Therefore, in 2019, Exactech decided to transition GXL liners out of the US market in favor of the XLE liner. Thus, at this time, the GXL liner has been transitioned entirely out of the US market. Bench testing reveals that Exactech’s new XLE liner does outperform the Connexion GXL liner in both volumetric wear and edge loading assessments.”
Over the course of 24 months prior to issuing the notification letters, Exactech made observations that the GXL polyethylene liners used as part of its hip replacement systems had risks of wear that could cause osteolysis or bone deterioration. In serious cases of wear and osteolysis, patients required revision surgery.
Similar to recall proceedings, Exactech has advised surgeons to encourage their patients who had GXL liners implanted within the last six years to come for an exam and x-rays to assess their hip replacement system for wear. Patients with symptoms including pain, stiffness, mobility issues or joint weakness should immediately contact their surgeon. Patients unsure if their hip replacement contains the Exactech Connexion GXL liner can contact their surgeon’s office for confirmation.
Calling this number connects you with a Drugwatch.com representative. We will direct you to one of our trusted legal partners for a free case review.
Drugwatch.com's trusted legal partners support the organization's mission to keep people safe from dangerous drugs and medical devices. For more information, visit our partners page.